Glycogen branching enzyme
Glycogen branching enzyme (GBE) is an essential enzyme in the process of glycogen biosynthesis. It plays a critical role in the formation of glycogen's branched structure, which is vital for the efficient storage and mobilization of glucose within the body. This enzyme is found in a variety of organisms, including humans, where it is encoded by the GBE1 gene.
Function[edit]
The primary function of the glycogen branching enzyme is to introduce α-1,6-linked branches into the glucan chain during the synthesis of glycogen. This is achieved by cleaving a 1,4-α-D-glucosidic link and transferring a block of oligoglucoside to a primary hydroxyl group at the C-6 position, creating a 1,6-α-D-glucosidic link. The introduction of these branches is crucial as it increases the solubility of glycogen and creates more terminal ends, which are the points of action for both glycogen synthase and glycogen phosphorylase. These enzymes are responsible for glycogen synthesis and degradation, respectively, and their activity is significantly enhanced by the branching structure.
Clinical Significance[edit]
Mutations in the GBE1 gene, which encodes the glycogen branching enzyme, can lead to a rare and often severe metabolic disorder known as Glycogen Storage Disease Type IV (GSD IV), also known as Andersen's disease. This condition is characterized by a deficiency in the activity of GBE, leading to the abnormal structure of glycogen, known as polyglucosan bodies. These abnormal glycogen molecules accumulate in various tissues, including the liver, muscles, and nervous system, leading to a range of symptoms from liver dysfunction to neuromuscular impairments.
Structure[edit]
The glycogen branching enzyme is a single polypeptide that contains multiple domains, including a catalytic core and several carbohydrate-binding modules. These domains facilitate the enzyme's interaction with glycogen and are essential for its branching activity. The structure of GBE has been elucidated through various techniques, including X-ray crystallography, revealing insights into its mechanism of action and how mutations may affect its function.
Genetics[edit]
The GBE1 gene is located on chromosome 3 in humans and contains multiple exons that encode the enzyme. Mutations in this gene can be inherited in an autosomal recessive manner, meaning that an individual must inherit two defective copies of the gene to manifest the disease. Genetic testing can identify carriers of GBE1 mutations and help in the diagnosis and management of Glycogen Storage Disease Type IV.
Treatment and Management[edit]
There is currently no cure for Glycogen Storage Disease Type IV, and treatment is primarily supportive and symptomatic. Management strategies may include dietary modifications to manage liver disease and physical therapy to address muscle weakness. In severe cases, liver transplantation may be considered. Ongoing research is focused on developing gene therapy and other novel treatments to correct the underlying genetic defect and improve outcomes for individuals with this condition.
See Also[edit]
This medical article is a stub. You can help WikiMD by expanding the page. |
-
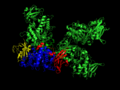 E. Coli Glycogen Branching Enzyme
E. Coli Glycogen Branching Enzyme -
 Glycogen Branching Enzyme Scheme
Glycogen Branching Enzyme Scheme

Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
