Primary ciliary dyskinesia
Rare autosomal recessive genetic disorder affecting ciliary function
| Primary ciliary dyskinesia | |
|---|---|
| Synonyms | PCD, Immotile cilia syndrome (outdated) |
| Pronounce | N/A |
| Specialty | N/A |
| Symptoms | Chronic respiratory infections, sinusitis, otitis media, bronchiectasis, infertility |
| Complications | Chronic lung disease, bronchiectasis, infertility, hearing loss |
| Onset | Early childhood |
| Duration | Lifelong |
| Types | N/A |
| Causes | Genetic mutations affecting ciliary function |
| Risks | Family history, autosomal recessive inheritance |
| Diagnosis | Genetic testing, nasal nitric oxide testing, ciliary biopsy, imaging studies |
| Differential diagnosis | Cystic fibrosis, Bronchiectasis, Asthma, Kartagener syndrome |
| Prevention | Genetic counseling |
| Treatment | Chest physiotherapy, antibiotics, supportive care, surgical interventions |
| Medication | Antibiotics, mucolytics |
| Prognosis | Variable; good management can improve quality of life |
| Frequency | Approximately 1 in 10,000 to 1 in 20,000 births |
| Deaths | N/A |
Primary ciliary dyskinesia (PCD) is a rare, ciliopathic autosomal recessive genetic disorder characterized by abnormal function of cilia, the microscopic hair-like structures lining various organs such as the respiratory tract, sinuses, Eustachian tube, middle ear, and reproductive organs. Historically termed "immotile cilia syndrome," the term is no longer preferred, as the cilia retain movement, although their action is typically inefficient or unsynchronized.
Pathophysiology[edit]
PCD is caused by genetic mutations that affect the structure and function of motile cilia. Normally, these cilia beat synchronously 7 to 22 times per second, facilitating mucus clearance through the mucociliary escalator. Dysfunctional cilia result in impaired mucus clearance, causing recurrent infections and chronic inflammation.
Signs and symptoms[edit]


The primary symptoms of PCD result from impaired mucus clearance and chronic respiratory infections. Symptoms usually begin early in childhood and can include:
- Chronic cough with excessive mucus production
- Recurrent sinusitis and nasal congestion
- Frequent ear infections (otitis media) leading to potential hearing loss
- Recurrent bronchitis and pneumonia
- Progressive development of bronchiectasis—permanent dilation and damage to the airways
- Reduced lung function over time
Non-respiratory symptoms include:
- Infertility due to impaired ciliary function in the reproductive tract:
Male infertility related to immotile sperm flagella Female infertility or increased risk of ectopic pregnancy due to impaired cilia in the fallopian tubes
Kartagener syndrome[edit]
Approximately 50% of patients with PCD have a subset of the condition known as Kartagener syndrome, characterized by the clinical triad of:
- Primary ciliary dyskinesia
- Situs inversus (mirror-image reversal of internal organs)
- Chronic sinusitis
Genetics[edit]
PCD is inherited in an autosomal recessive manner, requiring two copies of the defective gene—one from each parent. Numerous genes have been implicated, including DNAI1, DNAH5, and others involved in the structure and function of ciliary dynein arms and associated proteins.
Diagnosis[edit]
Diagnosing PCD can be challenging due to overlapping symptoms with other respiratory conditions such as asthma, bronchiectasis, and cystic fibrosis. Diagnostic methods include:
- Clinical evaluation: Chronic respiratory symptoms starting early in childhood.
- Nasal nitric oxide testing: Levels typically very low in patients with PCD.
- Genetic testing: Identifies specific genetic mutations associated with PCD.
- High-speed videomicroscopy: Examination of ciliary beat frequency and coordination from nasal or bronchial biopsy samples.
- Electron microscopy: Detects structural abnormalities in cilia.
- Imaging: Chest X-ray and CT scans may demonstrate characteristic findings like bronchiectasis or mucus plugging.
Differential diagnosis[edit]
Conditions presenting similarly to PCD include:
- Cystic fibrosis
- Bronchiectasis from other causes
- Severe, chronic asthma
- Immunodeficiency disorders
- Allergic bronchopulmonary aspergillosis (ABPA)
Management and treatment[edit]
PCD has no cure, but management focuses on preventing complications and reducing symptom severity:
Chest physiotherapy[edit]
Daily airway clearance techniques help remove mucus, reducing infection frequency and preserving lung function:
- Postural drainage
- Chest percussion and vibration
- Active breathing techniques
- Use of mechanical mucus clearance devices
Medication[edit]
Pharmacologic management includes:
- Antibiotics for frequent respiratory infections, including prophylactic or intermittent courses.
- Mucolytics to decrease mucus viscosity (e.g., hypertonic saline inhalation).
- Bronchodilators, if airway hyperreactivity is present.
Surgical intervention[edit]
Surgery may be necessary for:
- Chronic sinusitis (e.g., endoscopic sinus surgery).
- Severe bronchiectasis complications.
- Placement of ventilation tubes in cases of chronic ear infections.
Fertility assistance[edit]
Patients experiencing infertility may benefit from assisted reproductive techniques such as in vitro fertilization (IVF).
Prognosis[edit]
PCD is a chronic, lifelong condition. Early diagnosis and aggressive management of infections and airway clearance can significantly improve quality of life and slow disease progression. Without appropriate management, PCD can result in severe lung damage and reduced lifespan.
Epidemiology[edit]
Primary ciliary dyskinesia is rare, affecting approximately 1 in 10,000 to 1 in 20,000 births globally. Prevalence may be higher in populations with increased consanguinity.
Research[edit]
Research continues to explore gene-specific therapies and advanced diagnostic techniques to improve outcomes and quality of life for individuals with PCD. Studies on nitric oxide pathways and ciliary biology provide insights into novel therapeutic approaches.
Gallery[edit]
-
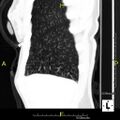 CT scan showing mucus impaction ("tree-in-bud")
CT scan showing mucus impaction ("tree-in-bud") -
 Bronchiectasis associated with Kartagener syndrome
Bronchiectasis associated with Kartagener syndrome

See also[edit]
External links[edit]
- Primary Ciliary Dyskinesia Foundation
- Genetic and Rare Diseases Information Center (GARD)
| Diseases of cilia | ||||||
|---|---|---|---|---|---|---|
See also: ciliary proteins
|


Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
