Cystinosis

Editor-In-Chief: Prab R Tumpati, MD
Obesity, Sleep & Internal medicine
Founder, WikiMD Wellnesspedia &
W8MD's weight loss doctor NYC
Philadelphia GLP-1 weight loss and GLP-1 clinic NYC
| Cystinosis | |
|---|---|

| |
| Synonyms | N/A |
| Pronounce | N/A |
| Specialty | N/A |
| Symptoms | Fanconi syndrome, growth retardation, photophobia, renal failure |
| Complications | Kidney failure, hypothyroidism, diabetes mellitus, muscle wasting |
| Onset | Infancy |
| Duration | Lifelong |
| Types | Nephropathic cystinosis, Intermediate cystinosis, Ocular cystinosis |
| Causes | Mutations in the CTNS gene |
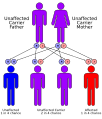
| Risks | Autosomal recessive inheritance |
| Diagnosis | Genetic testing, corneal cystine crystal examination |
| Differential diagnosis | Fanconi syndrome, cystinuria, other metabolic disorders |
| Prevention | N/A |
| Treatment | Cysteamine, kidney transplantation, symptomatic treatment |
| Medication | Cysteamine |
| Prognosis | Variable, depends on treatment |
| Frequency | 1 in 100,000 to 200,000 live births |
| Deaths | N/A |
Cystinosis
Cystinosis is a rare, inherited metabolic disorder characterized by the accumulation of the amino acid cystine within lysosomes, leading to cellular damage. This condition primarily affects the kidneys and eyes, but can also impact other organs and tissues.
Pathophysiology[edit]
Cystinosis is caused by mutations in the CTNS gene, which encodes the protein cystinosin. Cystinosin is responsible for transporting cystine out of lysosomes. When this transport is impaired, cystine accumulates, forming crystals that can cause cellular dysfunction and damage.
Types of Cystinosis[edit]
Cystinosis is classified into three main types based on the age of onset and severity of symptoms:
Nephropathic Cystinosis[edit]
This is the most common and severe form, typically presenting in infancy. It is characterized by renal tubular Fanconi syndrome, leading to excessive loss of water, sodium, potassium, and other substances in the urine.
Intermediate Cystinosis[edit]
Also known as juvenile or adolescent cystinosis, this form presents later in childhood or adolescence. It is less severe than nephropathic cystinosis but can still lead to significant kidney damage over time.
Ocular Cystinosis[edit]
This form primarily affects the eyes, causing photophobia and corneal crystal deposits. It usually presents in adulthood and does not typically involve kidney dysfunction.
Clinical Manifestations[edit]
Renal Symptoms[edit]
- Fanconi Syndrome: Characterized by polyuria, polydipsia, and growth retardation due to renal tubular dysfunction. - Chronic Kidney Disease: Progressive renal impairment leading to end-stage renal disease if untreated.
Ocular Symptoms[edit]
- Photophobia: Sensitivity to light due to corneal cystine crystal deposits. - Corneal Crystals: Visible deposits in the cornea that can be detected by slit-lamp examination.
Other Symptoms[edit]
- Hypothyroidism: Due to cystine accumulation in the thyroid gland. - Diabetes Mellitus: Resulting from pancreatic involvement. - Muscle Weakness: Due to cystine accumulation in muscle tissue.
Diagnosis[edit]
Diagnosis of cystinosis is based on clinical presentation, family history, and laboratory tests. Measurement of cystine levels in leukocytes is a key diagnostic test. Genetic testing can confirm mutations in the CTNS gene.
Treatment[edit]
Cysteamine Therapy[edit]
Cysteamine is the primary treatment for cystinosis. It reduces cystine accumulation by converting cystine into cysteine and cysteine-cysteamine mixed disulfide, which can exit the lysosome.
Supportive Care[edit]
- Renal Replacement Therapy: Dialysis or kidney transplantation for end-stage renal disease. - Electrolyte Replacement: To manage Fanconi syndrome. - Ocular Drops: Cysteamine eye drops to reduce corneal crystal deposits.
Prognosis[edit]
With early diagnosis and treatment, individuals with cystinosis can have improved outcomes. However, untreated cystinosis can lead to significant morbidity and mortality due to renal failure and other complications.
Gallery[edit]
-
 Cystinosis
Cystinosis -
 Cystinosis
Cystinosis


See Also[edit]
- Lysosomal storage disease - Fanconi syndrome - End-stage renal disease
Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
