Hemoglobin E

Editor-In-Chief: Prab R Tumpati, MD
Obesity, Sleep & Internal medicine
Founder, WikiMD Wellnesspedia &
W8MD's weight loss doctor NYC
Philadelphia GLP-1 weight loss and GLP-1 clinic NYC
| Hemoglobin E | |
|---|---|
_1yvt.png)
| |
| Synonyms | N/A |
| Pronounce | N/A |
| Specialty | N/A |
| Symptoms | Mild anemia, fatigue, splenomegaly |
| Complications | Hemolytic anemia, thalassemia |
| Onset | Birth |
| Duration | Lifelong |
| Types | Hemoglobinopathy |
| Causes | Genetic mutation in the HBB gene |
| Risks | Southeast Asian descent |
| Diagnosis | Blood test, hemoglobin electrophoresis |
| Differential diagnosis | Thalassemia, Iron deficiency anemia |
| Prevention | Genetic counseling |
| Treatment | Folic acid supplementation, blood transfusion |
| Medication | N/A |
| Prognosis | N/A |
| Frequency | Common in Southeast Asia |
| Deaths | N/A |


Hemoglobin E (HbE) is an abnormal form of hemoglobin, a protein found in red blood cells that carries oxygen throughout the body. This variant of hemoglobin is most commonly found in individuals of Southeast Asian descent, including those from Thailand, Cambodia, and Laos. Hemoglobin E is caused by a mutation in the beta globin gene, which is one of the two genes that encode the protein subunits of hemoglobin.
Structure and Function[edit]
Hemoglobin E is structurally similar to normal hemoglobin, but it has a single amino acid substitution in the beta globin chain. This substitution results in a change in the protein's structure, which can affect its function. Despite this, individuals with hemoglobin E typically do not experience any symptoms or health problems related to the mutation.
Genetics[edit]
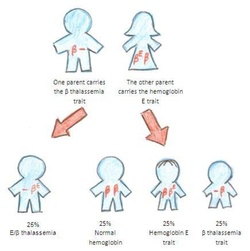
Hemoglobin E is inherited in an autosomal recessive manner, meaning that an individual must inherit two copies of the mutated gene (one from each parent) to have the condition. Those who inherit only one copy of the mutated gene are carriers and typically do not experience any health problems.
Clinical Significance[edit]
While individuals with hemoglobin E typically do not experience any symptoms, those who inherit two different abnormal hemoglobin genes (one for hemoglobin E and one for another variant, such as hemoglobin S or beta thalassemia) can have a more severe condition known as Hemoglobin E/beta thalassemia. This condition can cause a range of symptoms, including anemia, enlarged spleen, and skeletal deformities.
Diagnosis and Treatment[edit]
Hemoglobin E can be diagnosed through a blood test that measures the types of hemoglobin present. Treatment is typically not necessary for individuals with hemoglobin E, unless they also have another hemoglobin disorder.
See Also[edit]
This medical article is a stub. You can help WikiMD by expanding the page. |
Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
