Thalassemia

Editor-In-Chief: Prab R Tumpati, MD
Obesity, Sleep & Internal medicine
Founder, WikiMD Wellnesspedia &
W8MD's weight loss doctor NYC
Philadelphia GLP-1 weight loss and GLP-1 clinic NYC
| Thalassemia | |
|---|---|

| |
| Synonyms | N/A |
| Pronounce | N/A |
| Specialty | N/A |
| Symptoms | Anemia, fatigue, pale skin, jaundice, bone deformities |
| Complications | Iron overload, heart failure, liver disease, infections |
| Onset | Childhood |
| Duration | Lifelong |
| Types | Alpha thalassemia, Beta thalassemia |
| Causes | Genetic mutation |
| Risks | Family history, certain ethnicities (Mediterranean, Middle Eastern, South Asian) |
| Diagnosis | Complete blood count, hemoglobin electrophoresis, genetic testing |
| Differential diagnosis | Iron-deficiency anemia, sickle cell disease |
| Prevention | Genetic counseling |
| Treatment | Blood transfusion, iron chelation therapy, bone marrow transplant |
| Medication | Deferoxamine, deferasirox, deferiprone |
| Prognosis | N/A |
| Frequency | Common in certain populations |
| Deaths | Varies depending on severity and treatment |
Other names[edit]
- Mediterranean anemia;
- Cooley anemia;
- Beta thalassemia;
- Alpha thalassemia
- Thalassemia is an inherited blood disorder that reduces the production of functional hemoglobin (the protein in red blood cells that carries oxygen).
- This causes a shortage of red blood cells and low levels of oxygen in the bloodstream, leading to a variety of health problems.
- Hemoglobin is the protein in red blood cells that carries oxygen.
- The disorder results in large numbers of red blood cells being destroyed, which leads to anemia.


Types[edit]
Hemoglobin is made of two proteins:
- Alpha globin
- Beta globin
Thalassemia occurs when there is a defect in a gene that helps control production of one of these proteins. There are two main types of thalassemia:
- Alpha thalassemia occurs when a gene or genes related to the alpha globin protein are missing or changed (mutated).
- Beta thalassemia occurs when similar gene defects affect production of the beta globin protein.
There are many forms of thalassemia. Each type has many different subtypes. Both alpha and beta thalassemia include the following two forms:
- Thalassemia major
- Thalassemia minor
You must inherit the gene defect from both parents to develop thalassemia major. Thalassemia minor occurs if you receive the faulty gene from only one parent. People with this form of the disorder are carriers of the disease. Most of the time, they do not have symptoms. Beta thalassemia major is also called Cooley anemia.
Epidemiology[edit]
Alpha thalassemias occur most often in people from Southeast Asia, the Middle East, China, and in those of African descent. Beta thalassemias occur most often in people of Mediterranean origin. To a lesser extent, Chinese, other Asians, and African Americans can be affected.
Risk factors[edit]
Risk factors for thalassemia include:
- Asian, Chinese, Mediterranean, or African American ethnicity
- Family history of the disorder
Cause and genetics[edit]
- Hemoglobin is made up of two different components (subunits): beta globin and alpha globin.
- The HBB gene provides instructions for making beta globin, while the HBA1 and HBA2 genes provide instructions for making alpha globin.
- Each person has two copies of each of these genes, one inherited from the mother and one from the father.
- Changes (mutations) in the HBB gene lead to reduced levels of beta globin and cause beta thalassemia.
- Loss (deletion) of some or all of the HBA1 and/or HBA2 genes results in a shortage of alpha globin, leading to alpha thalassemia.
Inheritance[edit]
- In general, thalassemia is inherited in an autosomal recessive manner; however, the inheritance can be quite complex as multiple genes can influence the production of hemoglobin.
Inheritance of beta thalassemia[edit]
- Most people affected by beta thalassemia have mutations in both copies of the HBB gene in each cell.
- The parents of an affected person usually each carry one mutated copy of the gene and are referred to as carriers.
- Carriers typically do not show signs or symptoms of the condition; although some carriers of beta thalassemia develop mild anemia.
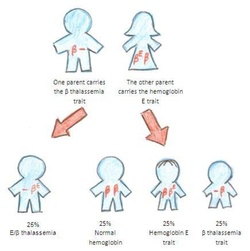
- When two carriers of an autosomal recessive condition have children, each child has a 25% (1 in 4) risk to have the condition, a 50% (1 in 2) risk to be a carrier like each of the parents, and a 25% chance to not have the condition and not be a carrier.
Alpha thalassemia[edit]
- The inheritance of alpha thalassemia is complicated by the fact that mutations in two different genes (HBA1 and HBA2) are associated with the condition.
- People have two copies of the HBA1 gene and two copies of the HBA2 gene in each cell.
- For each gene, one copy is inherited from the mother and one is inherited from the father.
- If each parent is missing at least one gene copy, their children are at risk for having alpha thalassemia.
- However, the exact risk and the severity of each child's condition depends on how many gene copies are lost (deleted) and which combination of the HBA1 and HBA2 genes are affected
Signs and symptoms[edit]
The signs and symptoms vary depending on the severity of the thalassemia. For example, people affected by milder forms of thalassemia can develop mild anemia or may have no signs or symptoms of the condition at all. Intermediate forms of thalassemia can cause mild to moderate anemia and may be associated with other health problems such as slowed growth, delayed puberty, bone problems and/or an enlarged spleen. In addition to the signs and symptoms seen in intermediate thalassemia, people with severe forms of thalassemia may also experience severe anemia, poor appetite, paleness, dark urine, yellow discoloration of skin (jaundice), and enlarged liver or heart.
Diagnosis[edit]
Your health care provider will do a physical exam to look for an enlarged spleen. A blood sample will be sent to a laboratory to be tested.
- Red blood cells will appear small and abnormally shaped when looked at under a microscope.
- A complete blood count (CBC) reveals anemia.
- A test called hemoglobin electrophoresis shows the presence of an abnormal form of hemoglobin.
- A test called mutational analysis can help detect alpha thalassemia.
- Yes, genetic testing is available for HBB, HBA1 and HBA2, the genes known to cause thalassemia. Carrier testing for at-risk relatives and prenatal testing are possible if the disease-causing mutations in the family are known.
Treatment[edit]
- Treatment for thalassemia major often involves regular blood transfusions and folate supplements.
- If you receive blood transfusions, you should not take iron supplements. Doing so can cause a high amount of iron to build up in the body, which can be harmful.
- People who receive a lot of blood transfusions need a treatment called chelation therapy.
- This is done to remove excess iron from the body.
- A bone marrow transplant may help treat the disease in some people, especially children.
Prognosis[edit]
- The long-term outlook (prognosis) for people with thalassemia depends on the type and severity of the condition.
- For example, severe thalassemia can cause early death due to heart failure, while less severe forms of thalassemia often do not shorten lifespan. Fortunately, improved treatment options have resulted in increased survival and better quality of life for people affected by moderate to severe thalassemia.
| Diseases of red blood cells | ||||
|---|---|---|---|---|
|
NIH genetic and rare disease info[edit]
Thalassemia is a rare disease.
| Rare and genetic diseases | ||||||
|---|---|---|---|---|---|---|
|
Rare diseases - Thalassemia
|

Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
