Kallmann syndrome
A genetic condition characterized by delayed or absent puberty and an impaired sense of smell
| Kallmann syndrome | |
|---|---|
| 19 year old Kallmann syndrome patient, pre-diagnosis.jpg | |
| Kallmann syndrome is characterized by a combination of hypogonadotropic hypogonadism and anosmia. | |
| Synonyms | Kallmann's hereditary anosmia, Hypogonadotropic hypogonadism with anosmia
|
| Specialty | Endocrinology, Medical genetics |
| Symptoms | Absent or delayed puberty, infertility, inability to smell, small testes or ovaries, delayed or absent secondary sexual characteristics |
| Complications | Osteoporosis, psychological impact due to delayed puberty or infertility |
| Usual onset | Present at birth |
| Duration | Lifelong |
| Types | X-linked, autosomal dominant, and autosomal recessive forms |
| Causes | Mutations in genes such as KAL1, FGFR1, PROKR2, and others affecting GnRH neuron migration |
| Risk factors | Family history of the syndrome; genetic mutations |
| Diagnosis | Physical examination, MRI of the brain, hormonal blood tests, olfactory testing, genetic testing |
| Differential diagnosis | Constitutional delay of growth and puberty, other forms of hypogonadotropic hypogonadism |
| Prevention | None |
| Treatment | Hormone replacement therapy, gonadotropin therapy, assisted reproductive technology for fertility |
| Medication | Testosterone (in males), estrogen and progesterone (in females), GnRH or gonadotropin injections |
| Prognosis | Generally good with appropriate hormone therapy; fertility may be restored in some cases |
| Frequency | 1:30,000 males, 1:125,000 females |
| Deaths | Rare, not typically life-threatening |

Kallmann syndrome is a rare genetic disorder that is characterized by a combination of hypogonadotropic hypogonadism and anosmia or hyposmia. It is a form of hypogonadotropic hypogonadism where the production of gonadotropin-releasing hormone (GnRH) is deficient, leading to a lack of sexual development and a diminished or absent sense of smell.
Pathophysiology[edit]
Kallmann syndrome is caused by a failure in the development of the olfactory bulbs and the migration of GnRH-producing neurons during embryonic development. This results in the absence or underdevelopment of the olfactory bulbs and a deficiency in GnRH, which is crucial for the stimulation of the pituitary gland to release luteinizing hormone (LH) and follicle-stimulating hormone (FSH). These hormones are essential for the normal function of the gonads and the onset of puberty.
Genetics[edit]
Kallmann syndrome can be inherited in an X-linked recessive, autosomal dominant, or autosomal recessive manner. Several genes have been implicated in the condition, including:
- KAL1 on the X chromosome, which is responsible for the X-linked form of the disorder.
- FGFR1 (also known as KAL2), which can cause an autosomal dominant form.
- PROKR2 and PROK2, which are associated with autosomal recessive forms.
Clinical Features[edit]
The primary clinical features of Kallmann syndrome include:
- Delayed or absent puberty
- Anosmia or hyposmia (reduced or absent sense of smell)
- Infertility due to hypogonadism
Additional features may include:
- Cleft lip or palate
- Hearing loss
- Renal agenesis (absence of one kidney)
- Mirror movements (bimanual synkinesis)
Diagnosis[edit]
Diagnosis of Kallmann syndrome is based on clinical evaluation, family history, and laboratory tests. Key diagnostic criteria include:
- Low levels of sex steroids (testosterone in males, estrogen in females)
- Low or normal levels of LH and FSH
- MRI imaging may reveal underdeveloped or absent olfactory bulbs
- Genetic testing can confirm mutations in known associated genes
Treatment[edit]
Treatment for Kallmann syndrome focuses on hormone replacement therapy to induce and maintain secondary sexual characteristics and fertility. Options include:
- Testosterone replacement therapy for males
- Estrogen and progesterone therapy for females
- Pulsatile GnRH therapy or gonadotropin injections to stimulate fertility
Prognosis[edit]
With appropriate treatment, individuals with Kallmann syndrome can achieve normal sexual development and fertility. However, the sense of smell typically does not improve with treatment.
Gallery[edit]
-
 Franz J. Kallmann, circa 1950
Franz J. Kallmann, circa 1950 -
 The structure of GNRH1
The structure of GNRH1 -
 Singer Jimmy Scott (r), whose unusual voice was due to Kallman syndrome
Singer Jimmy Scott (r), whose unusual voice was due to Kallman syndrome -
 Testosterone gel sachets, Testosterone undecanoate injection (Nebido), Human chorionic gonadotropin (hCG) injection, Menotropin injection (hMG).
Testosterone gel sachets, Testosterone undecanoate injection (Nebido), Human chorionic gonadotropin (hCG) injection, Menotropin injection (hMG). -
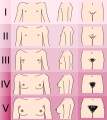 Tanner scale-female
Tanner scale-female -
 The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism
The genetic and molecular basis of idiopathic hypogonadotropic hypogonadism






Related pages[edit]
- Hypogonadotropic hypogonadism
- Anosmia
- Gonadotropin-releasing hormone
- Luteinizing hormone
- Follicle-stimulating hormone
External links[edit]
| X-linked disorders |
|---|
|
|

Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
