Fabry disease

Editor-In-Chief: Prab R Tumpati, MD
Obesity, Sleep & Internal medicine
Founder, WikiMD Wellnesspedia &
W8MD's weight loss doctor NYC
Philadelphia GLP-1 weight loss and GLP-1 clinic NYC
| Fabry disease | |
|---|---|

| |
| Synonyms | Anderson-Fabry disease, alpha-galactosidase A deficiency |
| Pronounce | |
| Specialty | Medical genetics |
| Symptoms | Pain, angiokeratoma, hypohidrosis, corneal opacity, tinnitus, hearing loss, kidney failure, heart disease |
| Complications | N/A |
| Onset | Childhood to adulthood |
| Duration | Lifelong |
| Types | N/A |
| Causes | Mutations in the GLA gene |
| Risks | Family history |
| Diagnosis | Genetic testing, enzyme assay |
| Differential diagnosis | Rheumatic fever, rheumatoid arthritis, multiple sclerosis |
| Prevention | N/A |
| Treatment | Enzyme replacement therapy, pain management, kidney transplantation |
| Medication | Agalsidase alfa, agalsidase beta, migalastat |
| Prognosis | Variable, depends on organ involvement and treatment |
| Frequency | 1 in 40,000 to 1 in 117,000 males |
| Deaths | |
Fabry disease is a rare genetic disorder that results from the buildup of a particular type of fat, called globotriaosylceramide, in the body's cells. It is an X-linked recessive disorder caused by mutations in the GLA gene, which leads to a deficiency of the enzyme alpha-galactosidase A.
Pathophysiology[edit]
Fabry disease is characterized by the accumulation of glycosphingolipids in the lysosomes of various cell types. The deficiency of alpha-galactosidase A enzyme activity results in the progressive deposition of globotriaosylceramide in the vascular endothelium, smooth muscle cells, and other tissues, leading to multi-systemic manifestations.
Clinical Manifestations[edit]
The symptoms of Fabry disease can vary widely among affected individuals, but common manifestations include:
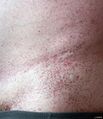
- Angiokeratomas: Small, dark red spots on the skin, often found in the "bathing trunk" area.
- Acroparesthesia: Episodes of pain and burning sensations in the hands and feet.
- Corneal verticillata: Whorled patterns on the cornea, detectable by an eye examination.
- Renal impairment: Progressive kidney damage leading to chronic kidney disease.
- Cardiac involvement: Hypertrophic cardiomyopathy, arrhythmias, and heart failure.
- Cerebrovascular disease: Increased risk of stroke and transient ischemic attacks.
Diagnosis[edit]
Diagnosis of Fabry disease is typically confirmed through:
- Enzyme assay: Measurement of alpha-galactosidase A activity in blood or skin fibroblasts.
- Genetic testing: Identification of mutations in the GLA gene.
- Biopsy: Histological examination of affected tissues may show characteristic lipid deposits.
Treatment[edit]
The management of Fabry disease includes:
- Enzyme replacement therapy (ERT): Intravenous administration of recombinant alpha-galactosidase A to reduce globotriaosylceramide accumulation.
- Chaperone therapy: Use of pharmacological chaperones to stabilize the mutant enzyme and enhance its activity.
- Symptomatic treatment: Pain management, renal protection, and cardiovascular care.
- Gene therapy: An emerging approach aimed at correcting the underlying genetic defect.
Prognosis[edit]
The prognosis of Fabry disease varies depending on the severity of organ involvement and the effectiveness of treatment. Early diagnosis and initiation of therapy can improve outcomes and quality of life.
Epidemiology[edit]
Fabry disease is estimated to affect approximately 1 in 40,000 to 1 in 117,000 live births. It is more common in males due to its X-linked inheritance pattern, but females can also be affected, often with milder symptoms.
Images[edit]
-
 Fabry disease
Fabry disease -
 Fabry disease
Fabry disease


See Also[edit]
Template:Lysosomal storage disorders
| Genetic disorders relating to deficiencies of transcription factor or coregulators | ||||||||||||||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
