Charcot–Marie–Tooth disease
| Charcot-Marie-Tooth disease | |
|---|---|
| Charcot-marie-tooth foot.jpg | |
| The foot of a person with Charcot–Marie–Tooth disease: The lack of muscle, a high arch, and claw toes are signs of this genetic disease. | |
| Synonyms | Charcot–Marie–Tooth neuropathy, peroneal muscular atrophy, Dejerine-Sottas Syndrome |
| Pronunciation | [ʃaʁko maʁi tuːθ]
|
| Symptoms | Foot drop, hammer toe, peripheral muscle wasting of lower legs
|
| Usual onset | Childhood - early adulthood |
| Duration | Lifelong
|
| Causes | Family history (genetics) |
| Risk factors | Family history (genetics), high-arched feet, flat-arched feet |
| Diagnosis | Genetic testing, nerve conduction study or electromyogram (EMG) |
| Differential diagnosis | Muscular dystrophy
|
| Treatment | management to maintain function
|
| Prognosis | progressive |
| Frequency | 1 in 2,500
|
Charcot-Marie-Tooth disease (CMT) is a hereditary motor and sensory neuropathy that affects the peripheral nervous system. It is characterized by the progressive loss of muscle tissue and touch sensation in various parts of the body. CMT is the most commonly inherited neurological disorder, impacting approximately one in 2,500 people.
The disease typically manifests in early childhood or early adulthood, although symptoms may also appear later in life. Foot drop, where the front of the foot cannot be lifted properly, is often the initial symptom. This can lead to the development of hammer toe, a condition where the toes remain curled. As the disease progresses, muscle wasting in the lower legs can result in a distinctive appearance known as "stork leg" or "inverted champagne bottle." Weakness in the hands and forearms may also occur over time.
Loss of touch sensation in the feet, ankles, legs, hands, wrists, and arms is common in various types of CMT. Painful spasmodic muscular contractions can occur intermittently, causing significant disability. High-arched feet (pes cavus) or flat-arched feet (pes planus) are often associated with the disorder. Sensory and proprioceptive nerves in the hands and feet are frequently affected, while unmyelinated pain nerves remain intact. Symptoms can be triggered or exacerbated by overuse of the affected limbs.
The progression and symptoms of CMT can vary from person to person. In addition to muscle and sensory issues, individuals with CMT may experience grinding of teeth (bruxism), squinting, breathing difficulties, hearing and vision problems, neck and shoulder muscle weakness, scoliosis (resulting in hunching and loss of height), malformed hip sockets, gastrointestinal issues, and difficulties with chewing, swallowing, and speaking. Tremors and exacerbation of symptoms during pregnancy or periods of severe emotional stress have also been reported.
CMT is caused by genetic mutations that affect neuronal proteins involved in nerve signal transmission and myelin sheath formation. Mutations in different genes can result in various types and subtypes of CMT. The duplication of a region on chromosome 17, including the PMP22 gene, is the most common cause of CMT. Mutations in the MFN2 gene on chromosome 1 can lead to mitochondrial dysfunction and disrupted axonal transport. X-linked CMT (CMTX) is caused by mutations in the GJB1 gene, which affects connexin 32, a gap junction protein involved in molecular exchange and signal transport in Schwann cells.
Diagnosis of CMT involves various tests, including nerve conduction studies to measure the speed of nerve impulses, nerve biopsies, and DNA testing. While DNA testing can provide a definitive diagnosis, not all genetic markers for CMT are currently known. A thorough evaluation by a neurologist or rehabilitation medicine specialist is necessary, considering symptoms, family history, and physical examinations that assess muscle weakness and sensory loss.
Unfortunately, there is currently no cure for CMT, and treatment focuses on managing symptoms and maintaining function. Physical therapies, assistive devices, corrective surgeries, and analgesic medications may be used to alleviate pain and improve mobility. Ongoing research aims to better understand the underlying mechanisms of CMT and develop targeted treatments for this complex disorder.
See also[edit]
- Charcot-Marie-Tooth disease classifications
- Palmoplantar keratoderma and spastic paraplegia
- Hereditary motor and sensory neuropathies
- Hereditary motor neuropathies
- Low copy repeats
- Christina's World

This WikiMD article can only be edited by registered and verified editors. You can log in or register.
| Inherited disorders of trafficking / vesicular transport proteins | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
See also vesicular transport proteins
|
| Cell membrane protein disorders (other than Cell surface receptor, enzymes, and cytoskeleton) | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|
See also other cell membrane proteins
|
| Deficiencies of intracellular signaling peptides and proteins | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
|
-
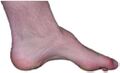 Foot affected by Charcot–Marie–Tooth disease
Foot affected by Charcot–Marie–Tooth disease -
 Chromosome 17
Chromosome 17 -
 Jean-Martin Charcot
Jean-Martin Charcot



Sponsored Health Resource

W8MD Weight Loss, Sleep & MedSpa
Looking for physician-supervised weight loss, semaglutide, tirzepatide, or GLP-1 receptor agonist options? W8MD helps eligible patients in New York City, Brooklyn, New Jersey, Connecticut, Pennsylvania, Delaware, and greater Philadelphia with medical weight loss, sleep medicine, and long-term maintenance support.
GLP-1 specials: Affordable GLP-1 injections NYC and Philadelphia starting from $29.99/week and up for semaglutide with insurance accepted for qualifying visits, and $45/week and up for tirzepatide with insurance accepted for qualifying visits. Self-pay options start from $59.99/week and up for semaglutide and $69.99/week and up for tirzepatide.
- Medical weight loss NYC
- Affordable GLP-1 injections NYC
- Budget GLP-1 weight loss shots Philadelphia
- New Jersey medical weight loss
- NYC medical weight loss blog
- Philadelphia weight loss blog
- Sleep medicine and sleep apnea services
- W8MD MedSpa and wellness
Book a W8MD appointment · View GLP-1 specials
Paid promotional message. Eligibility, pricing, insurance coverage, medication availability, and results vary. Medical evaluation required.

Medical Disclaimer: WikiMD is for informational purposes only and is not a substitute for professional medical advice. Content may be inaccurate or outdated and should not be used for diagnosis or treatment. Always consult your healthcare provider for medical decisions. Verify information with trusted sources such as CDC.gov and NIH.gov. By using this site, you agree that WikiMD is not liable for any outcomes related to its content. See full disclaimer.
Credits:Most images are courtesy of Wikimedia commons, and templates, categories Wikipedia, licensed under CC BY SA or similar.
Translate page: - East Asian
中文,
日本,
한국어,
South Asian
हिन्दी,
தமிழ்,
తెలుగు,
Urdu,
ಕನ್ನಡ,
Southeast Asian
Indonesian,
Vietnamese,
Thai,
မြန်မာဘာသာ,
বাংলা
European
español,
Deutsch,
français,
Greek,
português do Brasil,
polski,
română,
русский,
Nederlands,
norsk,
svenska,
suomi,
Italian
Middle Eastern & African
عربى,
Turkish,
Persian,
Hebrew,
Afrikaans,
isiZulu,
Kiswahili,
Other
Bulgarian,
Hungarian,
Czech,
Swedish,
മലയാളം,
मराठी,
ਪੰਜਾਬੀ,
ગુજરાતી,
Portuguese,
Ukrainian
